The Journal of Physical Chemistry C– Peter N. Njoroge,Bjørn Gading Solemsli, Asanka Wijerathne, Izar Capel Berdiell, Agnieszka Seremak, Mario Chiesa, Yu-Kai Liao, Beatrice Garetto, Nishant Patel, Karoline Kvande, Elisa Borfecchia, Christopher Paolucci, Unni Olsbye, Pablo Beato, Stian Svelle and Sebastian Prodinge
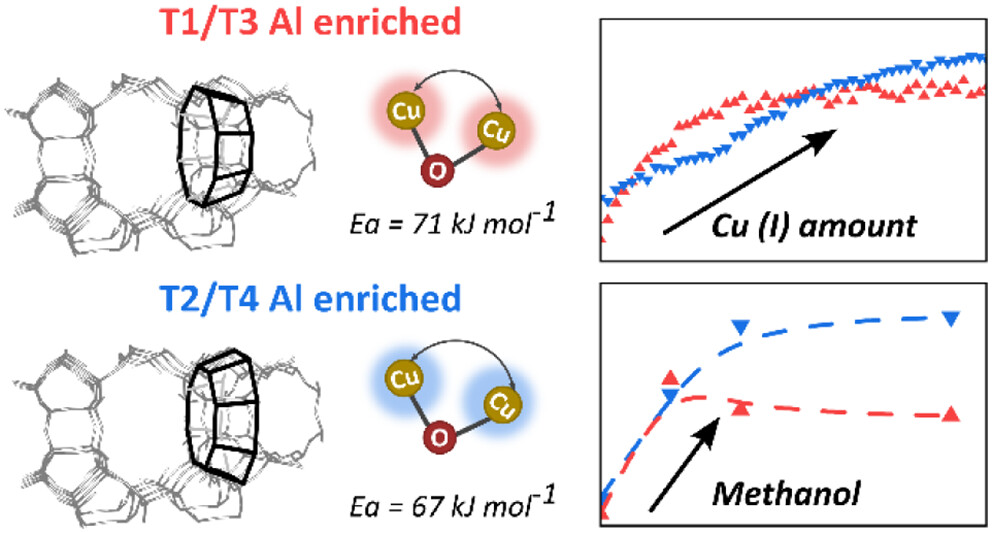
A series of copper-mordenite (MOR) samples of different provenances were investigated in the methane-tomethanol (MTM) reaction after preparing their copper-exchanged analogues. Noticeable activity improvements were observed when biasing the Al framework distribution of the confined side-pocket toward 12-ring openings (T2 and T4 enrichment) over 8-ring openings (T1 and T3), achieved by using K+ or Na+ in the synthesis gel, respectively. This was rationalized by performing a geometry optimization algorithm using density functional theory (DFT) simulations, which revealed distortions in the structure of
the pores among different idealized zeolite models. From this, effects on the copper species were observed, as evidenced from
both electron paramagnetic resonance (EPR) spectroscopy and operando X-ray absorption spectroscopy (XAS), which suggested varying monomeric [Cu]2+/[CuOH]+ concentrations with intrinsic copper reducibility differences. Monte Carlo simulations on selected MOR structures of the experimental series exposed dimeric structures with more acute Cu−O−Cu angles, thereby suggesting a more reactive system for Cu-MOR based on K+ in the synthesis gel, in line with the experimental finding. The combined insights from simulations, calculations, and experiments have enabled us to establish a synthesis-structure−activity relationship for mordenite in methane conversion, highlighting the reactive interplay between pore geometry and copper speciation.
Click here for the complete article!